Тики и синдром Туретта
Важным отличием тиков от других расстройств движения является возможность ненадолго подавить его, хотя это сопровождается нарастанием внутреннего напряжения. Кроме того, тикам может предшествовать ощущение непреодолимой потребности совершить движение, из-за чего может создаться впечатление, что оно произвольно.
Тики усиливается на фоне тревоги, волнения, недосыпания, стресса и уменьшаются при напряженной увлекательной для человека деятельности.
Как правило, тики возникают еще в детстве, максимально выражены в подростковом возрасте, а затем уменьшаются. Примерно у 1/3 пациентов с наступлением зрелого возраста тики могут полностью исчезнуть, вместе с тем в еще 1/3 случаев выраженность тиков может не уменьшаться даже во взрослом возрасте.
Кроме тиков пациентов также беспокоят психоэмоциональные нарушения. Это может быть обсессивно-компульсивный синдром, синдром нарушения внимания и гиперактивности, раздражительность, тревожность и др. Иногда подобные нарушения могут инвалидизировать больше, чем сами тики.
У меня тики, значит ли это, что у меня синдром Туретта?
Нет. Не у всех людей с тиками наблюдается синдром Туретта. Это могут быть как моторные или вокальные тики вне синдрома Туретта, так и другие состояния, при которых тики являются только одним из проявлений.
Для того чтобы установить причину тиков, необходима консультация специалиста. Он сможет подсказать, действительно ли ваши симптомы являются проявлением того или иного заболевания и при необходимости назначит дополнительные диагностические процедуры.
Какие есть возможности лечения?
Полностью вылечить заболевание невозможно. Но существует терапия, которая может улучшить психоэмоциональное состояние и уменьшить двигательные нарушения. Выраженность тиков при данном заболевании, а также то, насколько они мешают в повседневной жизни, сильно варьирует от случая к случаю. Поэтому терапия всегда индивидуальная. При легких проявлениях заболевания рекомендуется психотерапия, медикаментозное лечение назначается только при большей выраженности симптомов. В наиболее тяжелых случаях, когда лекарства не помогают, существует возможность оперативного лечения.
Для чего нужна консультация невролога?
Если вы подозреваете появление тиков у себя или своих близких, лучше сразу обратиться к специалисту. Он поможет вам установить правильный диагноз и подробно расскажет о заболевании.
Диагноз тиков и синдром Туретта является клиническим, и специальных тестов для его подтверждения не существует. Однако при нетипичных проявлениях и подозрении на вторичные причины врач может назначить вам доосбледование.
Невролог поможет вам определиться с необходимостью медикаментозной терапии и подберет необходимый круг препаратов с учетом всех проявлений заболевания.
При тяжелой степени выраженности тиков и отсутствии эффекта от трех и более препаратов, врач обсудит с вами возможности хирургического лечения.
Публикации в СМИ
Синдром амнестический
Амнестический синдром — психическое расстройство, возникающее вследствие органического поражения головного мозга и характеризующееся грубыми расстройствами памяти при отсутствии интеллектуальных нарушений.
Этиология. Амнестический синдром наиболее часто отмечают при недостаточности витамина B1 (чаще наблюдают при алкоголизме). Расстройство также может быть вызвано отравлением угарным газом, сосудистыми заболеваниями головного мозга, энцефалитами и опухолями третьего желудочка. Амнестический синдром может быть обусловлен любым процессом, вследствие которого происходит повреждение диэнцефальных и медиальных височных структур (например сосцевидных тел, гиппокампа, свода головного мозга).
Классификация и клиническая картина
• Амнестический синдром, обусловленный соматическим и/или неврологическим заболеванием •• Расстройства памяти (антероградная, ретроградная, фиксационная амнезии, конфабуляции), выявляющиеся не только на фоне делирия или деменции •• Дезориентация в месте, времени и собственной личности •• Снижение критических способностей •• Эмоционально-волевые расстройства (апатия, эмоциональная уплощённость, отсутствие инициативы) •• Расстройство непосредственно связано с соматическим и/или неврологическим заболеванием.
• Корсаковский психоз •• Расстройство вызвано недостаточностью витаминов группы В, особенно тиамина •• Прогноз неблагоприятен при развитии энцефалопатии Вернике, основные проявления которой — делирий, нистагм, офтальмоплегия, атаксия •• Течение обычно подострое, возможно острое или хроническое течение.
Дифференциальная диагностика направлена на выявление причины амнезии • Системные соматические заболевания •• Гипогликемия • Первичное заболевание (поражение) головного мозга •• Эпилепсия височная •• ЧМТ •• Опухоли •• Цереброваскулярные заболевания (атеросклероз, инсульты) •• Хирургические вмешательства •• Гипоксия (включая гипоксию при попытке повешения или отравлении угарным газом) •• Рассеянный склероз •• Герпетический энцефалит • Интоксикации: окись углерода, изониазид, мышьяк, свинец • Психотропные вещества •• Алкоголь •• Нейротоксины •• Седативные средства • Психические расстройства •• Деменция •• Делирий •• Психогенная амнезия.
Лечение. Следует определить причину расстройства и при возможности устранить её, например при опухолях головного мозга показано оперативное вмешательство, при амнестическом синдроме, вызванном недостаточностью витамина В1, рекомендовано назначение больших доз витамина тиамина. Во всех других случаях, когда невозможно устранить причину расстройства, отсутствует специфическое лечение, тактика ведения пациентов аналогична таковой при деменции. Лекарства, улучшающие память, неэффективны.
Течение и прогноз. В большинстве случаев наблюдают хроническое течение. Прогноз более благоприятен при недостаточности витамина В1 при условии, если тут же начать лечение.
Синонимы • Корсаковский синдром • Корсаковский психоз • Алкогольный амнестический синдром
МКБ-10 • F04 Органический амнестический синдром, не вызванный алкоголем или другими психоактивными веществами
Код вставки на сайт
Синдром амнестический
Амнестический синдром — психическое расстройство, возникающее вследствие органического поражения головного мозга и характеризующееся грубыми расстройствами памяти при отсутствии интеллектуальных нарушений.
Этиология. Амнестический синдром наиболее часто отмечают при недостаточности витамина B1 (чаще наблюдают при алкоголизме). Расстройство также может быть вызвано отравлением угарным газом, сосудистыми заболеваниями головного мозга, энцефалитами и опухолями третьего желудочка. Амнестический синдром может быть обусловлен любым процессом, вследствие которого происходит повреждение диэнцефальных и медиальных височных структур (например сосцевидных тел, гиппокампа, свода головного мозга).
Классификация и клиническая картина
• Амнестический синдром, обусловленный соматическим и/или неврологическим заболеванием •• Расстройства памяти (антероградная, ретроградная, фиксационная амнезии, конфабуляции), выявляющиеся не только на фоне делирия или деменции •• Дезориентация в месте, времени и собственной личности •• Снижение критических способностей •• Эмоционально-волевые расстройства (апатия, эмоциональная уплощённость, отсутствие инициативы) •• Расстройство непосредственно связано с соматическим и/или неврологическим заболеванием.
• Корсаковский психоз •• Расстройство вызвано недостаточностью витаминов группы В, особенно тиамина •• Прогноз неблагоприятен при развитии энцефалопатии Вернике, основные проявления которой — делирий, нистагм, офтальмоплегия, атаксия •• Течение обычно подострое, возможно острое или хроническое течение.
Дифференциальная диагностика направлена на выявление причины амнезии • Системные соматические заболевания •• Гипогликемия • Первичное заболевание (поражение) головного мозга •• Эпилепсия височная •• ЧМТ •• Опухоли •• Цереброваскулярные заболевания (атеросклероз, инсульты) •• Хирургические вмешательства •• Гипоксия (включая гипоксию при попытке повешения или отравлении угарным газом) •• Рассеянный склероз •• Герпетический энцефалит • Интоксикации: окись углерода, изониазид, мышьяк, свинец • Психотропные вещества •• Алкоголь •• Нейротоксины •• Седативные средства • Психические расстройства •• Деменция •• Делирий •• Психогенная амнезия.
Лечение. Следует определить причину расстройства и при возможности устранить её, например при опухолях головного мозга показано оперативное вмешательство, при амнестическом синдроме, вызванном недостаточностью витамина В1, рекомендовано назначение больших доз витамина тиамина. Во всех других случаях, когда невозможно устранить причину расстройства, отсутствует специфическое лечение, тактика ведения пациентов аналогична таковой при деменции. Лекарства, улучшающие память, неэффективны.
Течение и прогноз. В большинстве случаев наблюдают хроническое течение. Прогноз более благоприятен при недостаточности витамина В1 при условии, если тут же начать лечение.
Синонимы • Корсаковский синдром • Корсаковский психоз • Алкогольный амнестический синдром
МКБ-10 • F04 Органический амнестический синдром, не вызванный алкоголем или другими психоактивными веществами
Синдром Гийена-Барре
Всегда ли синдром Гийена-Барре протекает тяжело?
СГБ протекает с разной степенью тяжести. Встречаются легкие и среднетяжелые формы, когда пациент сохраняет способность ходить. Это характерно для большинства пациентов. Тяжелые и крайне тяжелые формы заболевания, при которых пациент обездвижен и нуждается в искусственной вентиляции легких из-за слабости дыхательной мускулатуры, отмечаются у каждого четвертого пациента.
Проведение ЭНМГ-исследования необходимо во всех случаях при подозрении на СГБ, поскольку данный метод позволяет не только подтвердить поражение периферических нервов, но и уточнить характер их повреждения, следовательно определить форму заболевания. Исследование пациентов в ранние сроки заболевания (когда симптоматика только нарастает и очень важно быстро поставить диагноз) имеет особенности, поэтому должно быть проведено хорошо подготовленным и опытным специалистом на миорафе высокого класса. Методологические ошибки и недостаточный объем данного исследования часто приводят к ошибочным диагнозам. Поэтому мы рекомендуем проведение ЭНМГ в нашем центре.
Патогенетическая терапия используются, прежде всего, с целью прерывания «аутоиммунной агрессии», развивающейся при СГБ. При этом предполагается достичь торможения дальнейшего развития заболевания, снизить длительность периода нарастания симптоматики, ускорить начало периода восстановления и добиться максимально полного выздоровления.
Глюкокортикостероиды при СГБ неэффективны и ухудшают прогноз!
К неспецифическим методам лечения относят симптоматическую терапию и реабилитацию. При этом восстановительное лечение является ключевым как в остром, так и в отдаленном периодах. Прием нейрометаболических, ноотропных препаратов, витаминов группы В при СГБ не рекомендован ни в остром периоде, ни в восстановительном, в связи с отсутствием доказательной базы.
ФГБНУ НЦН уже многие годы занимается диагностикой и лечением пациентов с синдромом Гийена-Барре. Коллективом ФГБНУ НЦН во главе с член-корреспондентом РАН Супоневой Н.А. подготовлены клинические рекомендации по ведению данной категории пациентов (в настоящий момент проходят утверждение).
Сотрудники центра заболеваний периферической нервной системы консультируют пациентов амбулаторно в рамках ОМС и на коммерческой основе.
ЗАПИСЬ НА ПРИЕМ И ЭНМГ/иЭМГ ПО МНОГОКАНАЛЬНОМУ ТЕЛЕФОНУ
+7 (495) 374-77-76
+7 (985) 931-60-24
Атаксия
Введение
«Ataxia» в дословном переводе с греческого языка обозначает «беспорядок». Однако наше современное понимание этого термина заключается в плохо координированных движениях, связанных, главным образом, с повреждением мозжечка и/или мозжечковых связей. В дополнение к мозжечковой атаксии (объясняющей большую часть случаев атаксий в клинической практике) существует также случаи так называемой сенситивной и вестибулярной атаксии, вызываемые соответственно повреждениями спинальных проприоцептивных путей и вестибулярной системы.
Клинические проявления различных типов атаксий
Мозжечковая атаксия
Клинически церебеллярная атаксия манифестирует неустойчивой и шаткой походкой с расширенной базой, а также дискоординацией и неуклюжестью движений, дизартрией (скандированной, отрывистой речью), дисметрией саккад и осцилляциями. Пациенты обычно стоят с широко отставленными стопами, при попытке поставить ноги ближе друг к другу они начинают раскачиваться или даже падают, из-за неустойчивого равновесия требуется поддержка или опора на окружающие предметы. Даже небольшие проявления атаксии ходьбы могут быть выявлены при так называемой тандемной ходьбе по прямой. Атаксия может быть генерализованной или преимущественно нарушать ходьбу, движения в руках, ногах, речь, движения глаз; может быть односторонней или вовлекать обе стороны. Атаксия часто сопровождается мышечной гипотония, замедленностью движений, интенционным тремором (тремор действия, усиливающийся по амплитуде при приближении к цели), нарушением контроля сложных многосуставных движений (асинергия), усиленными постуральными рефлексами, нистагмом (обычно горизонтальным при мозжечковой атаксии) и некоторыми когнитивными и аффективными изменениями (так называемым «мозжечковым когнитивно-аффективным синдромом», вызываемым обычно острыми, достаточно большими ишемическими повреждениями задней доли мозжечка). Следует подчеркнуть, что двигательные нарушения при атаксии обычно не связаны с мышечной слабостью, гиперкинезами, спастичностью и т.д., однако, все они, а также и другие дополнительные симптомы могут усложнять клиническую картину заболевания. В свою очередь выраженная атаксия может быть основной причиной инвалидизации и социальной дезадаптации.
Сенситивная атаксия
По сравнению с мозжечковой сенситивная атаксия достаточно редка. Обычно она является следствием поражения задних столбов и, соответственно, нарушения проприоцептивной афферентации (например, при болезни Фридрейха, дефиците витаминов Е и В12, нейросифилисе). Сенситивная атаксия может быть диагностирована по отчетливому проприоцептивному дефициту и значительному усилению симптоматики при закрытии глаз. Иногда в таких случаях можно заметить феномен «псевдоатетоза» в пораженной конечности.
Вестибулярная атаксия
Вестибулярная дисфункция может вызывать синдром, обозначаемый «вестибулярная» (или «лабиринтная») атаксия. Фактически этот синдром можно считать определенным подтипом сенситивной атаксии. Пациенты с вестибулярной атаксией демонстрируют грубые нарушения ходьбы и стояния (вестибулярное нарушение равновесия), но без вовлечения конечностей и речи. При односторонних поражениях лабиринта значительно нарушена «фланговая походка» в сторону повреждения. Этот тип атаксии часто сопровождается головокружением, рвотой и потерей слуха
Патофизиология
Патофизиологически мозжечковая атаксия представляет собой несостоятельность нормальных анти-инерционных механизмов, которые отвечают за плавность, равномерность и точность движений
Атактические расстройства при поражениях мозжечка
Поражения мозжечка и мозжечковых путей могут быть обусловлены острой или хронической патологией (см. таблицу).
Острая атаксия
Острая атаксия обычно наблюдается при ишемическом (лакунарном, кардиоэмболическом и атеротромботическом инфаркте) или геморрагичеком инсульте, поражающем полушария мозжечка. Также она может наблюдаться при рассеянном склерозе, черепно-мозговой травме, инфекционном церебеллите или абсцессе мозжечка, паразитарной инвазии, синдроме MELAS, острых лекарственных интоксикациях и отравлениях (этанолом, нейролептиками, антиконвульсантами), аномалии Арнольда-Киари и других патологиях. В этих случаях атаксия часто ассоциирована с головной болью, рвотой, головокружением, симптомами поражения ствола и черепных нервов. Следует помнить, что даже небольшие инфаркты мозжечка и кровоизлияния в связи с ограниченным объемом задней черепной ямки – это потенциально жизнеугрожающие состояния, которые могут приводить к обструктивной гидроцефалии. Поэтому всем пациентам с остро развившейся мозжечковой атаксией необходимо экстренно проводить нейровизуализацию (КТ или МРТ) и при необходимости последующее вентрикулярное дренирование и/или декомпрессионную трепанацию задней черепной ямки. Эти же мероприятия рекомендованы при любых заболеваниях, сопровождающихся большими острыми повреждениями мозжечка с быстро прогрессирующим отеком структур задней черепной ямки. Что же касается люмбальной пункции у этих пациентов, то она строго противопоказана в виду риска вклинения.
Повторяющиеся пароксизмы острой атаксии наблюдаются при периодических (эпизодических) атаксиях. Эти наследственные заболевания вызваны генетическими дефектами ионных каналов (кальциевых, калиевых), которые в свою очередь приводят к нарушениям возбудимости нейронов. Некоторые пациенты с атактическими пароксизмами могут хорошо отвечать на прием ацетазоламида (ацетазоламид-чувствительные формы периодических атаксий). Периодические атаксии принадлежат к группе так называемых каналопатий.
Хроническая атаксия
Хроническая атаксия может быть вызвана рядом различных заболеваний (см. таблицу) как генетической, так и негенетической природы. Хроническая или подострая мозжечковая атаксия, особенно в молодом возрасте, является типичной манифестацией рассеянного склероза, диагноз которого подтверждается ремитирующим течением и множественными очагами демиелинизации в головном и спинном мозге на МРТ. Следует всегда помнить, что хроническая или подострая мозжечковая атаксия может вызываться опухолью (среди характерных для мозжечка опухолей – церебеллопонтинная шваннома, медуллобластома и гемангиобластома), нормотензивной гидроцефалией (синдром Хакими-Адамса) и паранеопластической мозжечковой дегенерацией (рак легких и другие системными новообразованиями); все эти заболевания требуют соответствующего и своевременного хирургического лечения. Дегенерация мозжечка также может быть вызвана хроническим алкоголизмом, гипотиреозом, глютеновой болезнью, дефицитом витамина В12, тепловым ударом, злоупотреблением некоторыми препаратами с анксиолитическим, снотворным и противосудорожным действием.
Хроническая прогрессирующая атаксия является ключевой особенностью дегенеративных атактических синдромов как наследственных, так и спорадических.
Наследственные атаксии – клинически и генетически гетерогенная группа заболеваний, передающихся чаще всего по аутосомно-доминантному или аутосомно-рецессивному типу.
Среди аутосомно-рецессивных и Х-сцепленных рецессивных атаксий наиболее часто встречается атаксия Фридрейха, вызываемая экспансией ГАА-повторов в некодируемом участке гена FRDA на хромосоме 9q. Белковый продукт этого гена, фратаксин, считается вовлеченным в гомеостаз митохондриального железа. Таким образом, болезнь Фридрейха представляет собой менделирующую форму митохондриальных цитопатий. Обычно заболевание манифестирует достаточно рано (до 20 лет) и проявляется смешанной сенситивно-мозжечковой атаксией, дизартрией, мышечной слабостью, кардиомиопатией, скелетными деформациями, диабетом и неуклонно прогрессирующим течением. Существует достаточно строгая корреляция между длиной экспансии и клиническими проявлениями болезни Фридрейха, так относительно позднее начало и «доброкачественное» течение характерно для непротяженной экспансии ГАА-повторов.
Диагноз
У пациентов с атактическими расстройствами диагноз основывается в первую очередь на нейровизуализационных (КТ, МРТ) и нейрофизиологических (вызванные потенциалы, электронейромиография и др.) исследованиях, которые предоставляют данные о структурных и функциональных характеристиках центральной и периферической нервной системы. В большинстве случаев наследственных атаксий сегодня доступна верификация диагноза с помощью ДНК-анализа как для самих больных, так и для их клинически здоровых родственников из группы «риска». Для предотвращения новых случаев заболевания в этих семьях может проводиться медико-генетическое консультирование и пренатальная ДНК-диагностика.
У пациентов со спорадическим вариантом атаксии необходим поиск всех возможных соматических расстройств, которые могут вызывать мозжечковую симптоматику (новообразования, эндокринные заболевания и др.). Атаксия может быть проявлением ряда метаболических заболеваний (см. таблицу), поэтому следует проводить соответствующий биохимический скрининг.
Лечение
Лечение и прогноз атактических синдромов основывается на их причине. При существовании радиального лечения (как например, хирургия опухолей мозжечка или коррекция дефицита витаминов) можно ожидать полного или частичного восстановления или, по крайней мере, прекращение дальнейшего прогрессирования.
Не существует лечения непосредственно самой атаксии. Ограниченный положительный эффект сообщался при дегенеративных атаксиях при приеме амантадина, буспирона, L-5-гидрокситриптофана, тиреотропин-релизинг-фактора и прегабалина, однако, эти данные не подтверждены рандомизированными исследованиями. Есть сообщения успешного лечения мозжечкового тремора изониазидом и некоторыми антиконвульсантами (клоназепамом, карбамазепином и топираматом); в некоторых случаях возможна стереотаксическая хирургия на ядрах таламуса.
Физиотерапия является важной составляющей в лечении пациентов с атаксией. Она направлена на предотвращение различных осложнений (таких как контрактуры и мышечные атрофии), поддержания физической формы, улучшения координации и ходьбы. Рекомендованы специальные комплексы «мозжечковых» и «сенсорных» упражнений, а также процедуры с биологической обратной связью и стабилографией.
На стадии разработки находятся первые подходы к генной и клеточной терапии наследственных атаксий; возможно, что именно эти технологии в будущем позволят совершить существенный прорыв в лечении.
Таблица. Причины острой и хронической атаксии
Острая атаксия
Хроническая атаксия
Острая лекарственная интоксикация и отравление:
MELAS, болезнь Лея и другие митохондриальные энцефаломиопатии с острым началом
Опухоли и мальформации с острой и подострой манифестацией
Дефицит тиамина (энцефалопатия Вернике)
Паранеопластическая мозжечковая дегенерация
Гипертермия (тепловой удар)
Наследственные болезни метаболизма:
Хроническая ишемия мозга
Нормотензивная гидроцефалия (синдром Хакима-Адамса)
Паранеопластическая дегенерация мозжечка
Мозжечковая дисплазия или гипоплазия (врожденная атаксия, обычно не прогрессирующая)
Прионные заболевания (атактическая форма)
Дефицит витамина B12
Гипертермия (тепловой удар)
Злоупотребление препаратами с анксиолитическим, снотворным и антиконвульсивным действием
Наследственные атаксия с аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным наследованием
Спорадические идиопатические дегенеративные атаксии:
Хромосомные нарушения
Хромосомные нарушения — это клинические синдромокомплексы, в основе которых лежат нарушения числа или структуры хромосом, то есть избыток или нехватка генетического материала, локализованного в той или иной хромосоме.
В норме у человека число хромосом равно 46, из которых 23 ребенок получает от матери и 23 аналогичные хромосомы от отца. В этом наборе гентического материала есть 2 особые хромосомы, которые были названы «половыми». Они определяют пол ребенка и ряд других важных признаков.
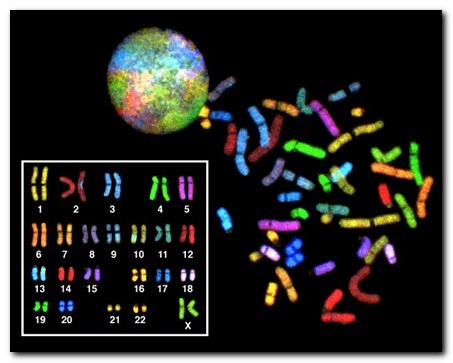
Таким образом, изменения числа хромосом (больше или меньше 46), а также изменение структуры хромосом (например, выпадение или удвоение даже небольшого кусочка хромосомы) получили название «хромосомные мутации».
Наиболее часто из них встречаются изменения модального числа хромосом — это отсутствие в хромосомном наборе какой-либо хромосомы (моносомия) или появление добавочной хромосомы (трисомия, тетрасомия и т.д.).
Число возможных изменений структуры хромосомы неисчислимое множество. К примеру, транслокации (обмен сегментами между разными хромосомами), делеции (выпадение участка хромосомы), дупликации (удвоение части хромосомы), инверсии (переворот сегмента хромосомы на 180 градусов) и т.д.
Хромосомные мутации, возникшие в половых клетках (сперматозоидах или яйцеклетках) или на первых этапах деления клеток зародыша, как правило, передаются большинству клеток развивающегося организма, вызывая множественные аномалии развития, а многие хромосомные изменения плода могут стать причиной спонтанных абортов и выкидышей, что важно учитывать в семьях, воспитывающих детей с задержками развития.
К факторам риска, способствующим их возникновению, относят ионизирующую радиацию, инфекции и интоксикации матери, эндокринные нарушения, психические травмы, воздействие ряда лекарственных препаратов и некоторых физиотерапевтических методов лечения.
Наиболее точно установлено, что причиной появления ребенка с хромосомными мутациями является не молодой возраст матерей (свыше 40 лет).
В последнее время очень большое значение придается факту скрытого носительства хромосомных нарушений у родителей родившегося ребенка (сбалансированные транслокации, мозаицизм). Изучение данного вопроса позволяет предотвратить риск повторного рождения ребенка с аналогичной формой заболевания.
Различают хромосомные синдромы, обусловленные изменением половых хромосом, и синдромы, вызванные аномалиями аутосом (любой из 44 неполовых хромосом).
Основными клиническими проявлениями аутосомных аномалий являются признаки психического и физического недоразвития, дисплазии (неправильное развитие), врожденные пороки развития (аномалии) и умственная отсталость различной степени тяжести. К врожденным порокам можно отнести: аномалии развития сердца, удвоение почки, расщелина неба, особенности строения кистей и стоп и многие другие. При заболеваниях, обусловленных нарушениями в системе половых хромосом, как правило, более характерны недоразвитие половых желез и аномалии развития вторичных половых признаков, также с симптомами задержки психо-речевого развития.
Различные хромосомные синдромы встречаются с разной частотой. По сводным данным многих исследований, распространенность наиболее частых из них среди новорожденных следующая:
трисомия по 21 хромосоме (синдром Дауна) 1:500
XXX (трисомия-Х) 1:1000 (девочек)
ХYY (синдром дубль-Y) 1:1000 (мальчиков)
ХХY (синдром Клайнфелтера) 1:1400 (мальчиков)
Х0 (синдром Шерешевского-Тернера) 1:3300 (девочек)
46,5р del (синдром «кошачьего крика») 1:4000
трисомия по 18 хромосоме (синдром Эдвардса) 1: 6800
трисомия по 13 хромосоме (синдром Патау) 1:7600

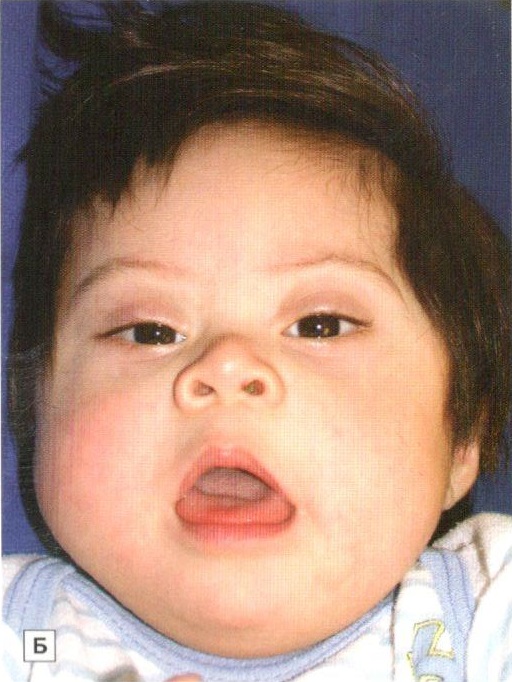
Установлено, что для синдрома Дауна характерно уменьшение размеров и веса головного мозга, а также аномалии развития мозга и мозговых сосудов. Отмечаются также структурные изменения в железах внутренней секреции, печени и сердце. Клиническая картина синдрома Дауна характеризуется проявлениями симптомов умственной отсталости. Характерен также и внешний вид таких больных: косо расположенные глазные щели, широкая уплощенная переносица, дополнительная кожная складка у внутреннего угла глаз, высокое стояние твердого неба (признаки эмбриональной задержки в развитии лицевого скелета), полуоткрытый рот, увеличенный высунутый язык с выраженными сосочками и глубокими бороздами (признаки дисфункции щитовидной железы), выпадение волос (дисфункция надпочечников), низкий рост, короткая шея, укороченные кисти и стопы, искривление мизинца, на ладонях имеется поперечная складка, на стопах увеличен промежуток между 1 и 2 пальцами, выражены внешние проявления гипогенитализма.
В клинической картине заболевания доминируют симптомы неврологической патологии, диффузная мышечная гипотония (снижение мышечного тонуса), благодаря чему больные гибки и иногда могут складываться как «перочинный ножик», расстройства координации движений, косоглазие, выраженные вегетососудистые нарушения.
Особенностью психического дефекта является относительная сохранность эмоциональной сферы по сравнению с тяжестью интеллектуального недоразвития. Так, больные ласковы, добродушны, послушны. Характерной особенностью таких детей является повышенная внушаемость, что является положительным фактором при проведении коррекционной работы и отрицательным при их развитии.
Уровень социального развития больных с синдромом Дауна зависит от степени и формы заболевания. Так, дети с более легкими формами умственной отсталости, хотя и медленно, но развиваютя, приобретая определенные навыки, знания, осваивая программу нескольких классов вспомогательной школы. Однако, как правило, большинство из них не достигают удовлетворительного уровня социальной адаптации и нуждаются в постоянной опеке. Им может быть оформлена инвалидность детства с момента точной диагностики заболевания. Особенностью возрастной динамики синдрома Дауна является позднее половое созревание и раннее появление признаков инволюции (25—30 лет). Мужчины с синдромом Дауна бесплодны, женщины могут давать потомство, половина которого также страдает синдромом Дауна.
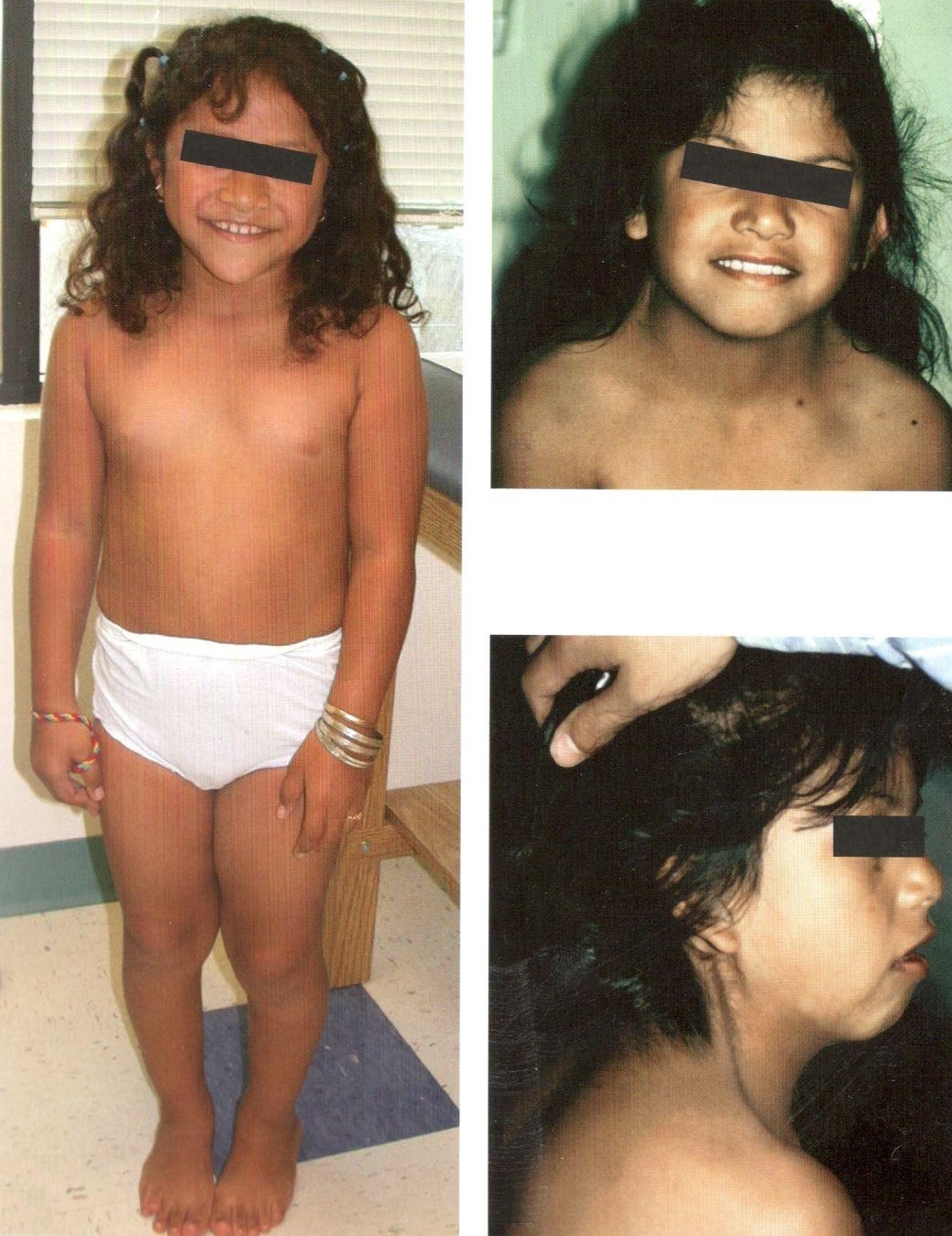
Впервые заболевание описано отечественным эндокринологом Н.А. Шерешевским (1925), а более подробно — американским эндокринологом Н. Тернером (N.H. Terner) л 1938 г. В основе заболевания лежит отсутствие одной хромосомы (половой Х-хромосомы) (45 вместо 46).
Клиническая картина синдрома характеризуется разной степенью умственной отсталости и ЗПРР, низким конечным ростом (135—145 см), замедлением полового развития, недоразвитием половых желез, аменореей, бесплодием и отсутствием грудных желез. Диспластические расстройства проявляются в виде короткой шеи и особых кожных складок, идущих от затылка к надплечью, укорочением 4 пальцев на руках и искривлением мизинцев, выраженной деформацией ушных раковин, наличием множественных пигментных родинок. Преимущественно данным синдромом страдают лица женского пола.
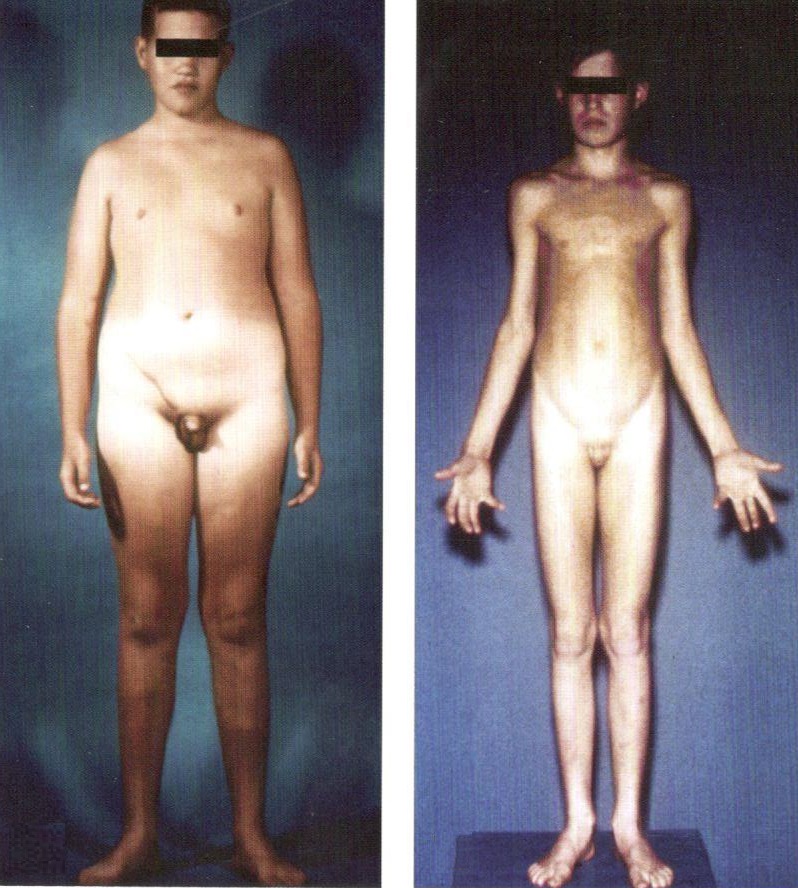
Клинические проявления синдрома Клайнфельтера варьируют от внешне нормального и интеллектуального развития до выраженного евнухоидизма и умеренной умственной отсталости. Однако в ряде случаев уже в раннем возрасте у больных отмечаются характерные своеобразные симптомы физического развития: низкий и узкий лоб, густые и жесткие волосы, высокое стояние таза, короткая, плоская и узкая грудная клетка, недоразвитие половых органов. Более отчетливо вышеперечисленные симптомы начинают обнаруживаться в подростковом, пубертатном возрасте. Характерен внешний вид взрослого больного с синдромом Клайнфельтера: высокий рост, астеническое сложение, узкие плечи, широкий таз, удлиненные конечности, слаборазвитая мускулатура, скудная растительность на лице и в подмышечных впадинах, ожирение и оволосение по женскому типу, сутулость, выраженные евнухоидные пропорции и гинекомастия (набухание грудных желез). Постоянными признаками синдрома Клайнфельтера являются недоразвитие половых органов и бесплодие.
Степень интеллектуального недоразвития у больных выражена тем глубже, чем больше дополнительных половых хромосом обнаруживается в кариотипе (46 или 49). Так, умеренная умственная отсталость зачастую приближается к психическому инфантилизму, что клинически проявляется недостаточностью внимания, восприятия, памяти, абстрактного мышления, чрезмерной внушаемостью, подражательностью, подчиняемостью, несамостоятельностью, чрезмерной привязанностью к близким, нередко с элементом назойливости. Глубокая незрелость эмоционально-волевой сферы проявляется в виде повышенного настроения, с эйфорическим оттенком, склонностью к эксплозивным аффективным вспышкам, неспособностью к длительному волевому усилию и напряженной деятельности. У больных, как правило, отсутствуют чувство долга и ответственности. При легких формах заболевания больные осознают свою неполноценность, что приводит к внутреннему конфликту и возникновению у них невротических реакций. Данным синдромом страдают лица мужского пола.
Синдром ломкой Х-хромосомы (Fragile X syndrome, FraХ). Начиная с 1980 года большое значение придают синдрому ломкой Х-хромосомы (Хq27.3) – именно с ним связывают развитие более чем 50 наследственных расстройств, включая ранний детский аутизм и 30% случаев умственной отсталости у мальчиков. Хрупкий участок Х-хромосомы впервые обнаружил Labs (1969).
Полная мутация в Х-хромосоме возникает только у женщин, и происходит это в процессе гаметогенеза, поэтому почти всегда страдают мальчики, получившие единственную Х-хромосому от матери. У девочек, получивших вторую Х-хромосому от отца, также могут быть нарушения развития, но они менее выражены, а тяжелые патологии встречаются много реже, чем у мальчиков. В отдельных случаях девочки могут получить обе ломкие хромосомы от матери, в этом случае частота и тяжесть патологии будет одинаковой с мальчиками.
Клиническую триаду синдрома ломкой Х-хромосомы образуют:
1) умеренная до степени тяжелой умственная отсталость. Лишь 30% лиц мужского пола имеют интеллект, стремящийся к нижней границе нормы, а среди женщин – носительниц такой хромосомной патологии примерно у 30% обнаруживаются признаки умственного недоразвития;
2) характерные особенности строения лица и черепа: выдающийся вперед высокий лоб, прогнатизм и удлиненные уши;
3) мальчики имеют увеличенные в размерах тестикулы (макроорхидизм).
Наблюдаются, кроме того, эпилептические припадки, синдром гиперактивности с дефицитом внимания, у более чем половины мальчиков аутизм и подобные аутизму расстройства, различные нарушения развития речи, персеверации, эхолалия, другие отклонения.
Женщины, унаследовавшие ломкую Х-хромосому с полной мутацией от своих матерей, могут быть склонны к развитию атипической депрессии, а также шизофреноподобного заболевания.
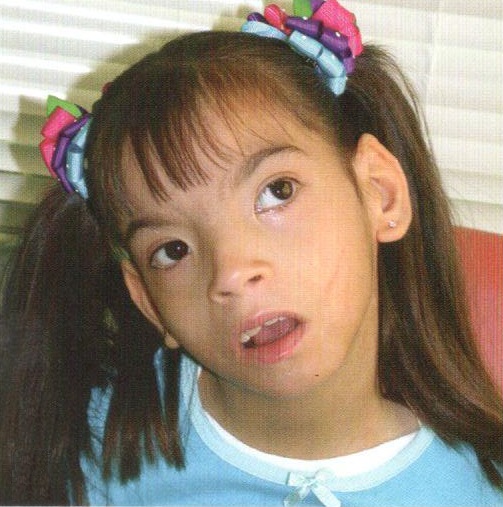
Синдром Вольфа—Хиршхорна.
В основе синдрома лежит изменение длины хромосомы из четвертой пары. Основные признаки заболевания у новорожденных: большое туловище, клювовидный нос и выступающее надпереносье, деформированные ушные раковины со складками, пучеглазие и колобома радужной оболочки (ее частичное отсутствие), общее недоразвитие во время беременности. Отмечается наличие четырех сгибательных складок на пальцах верхних конечностей.
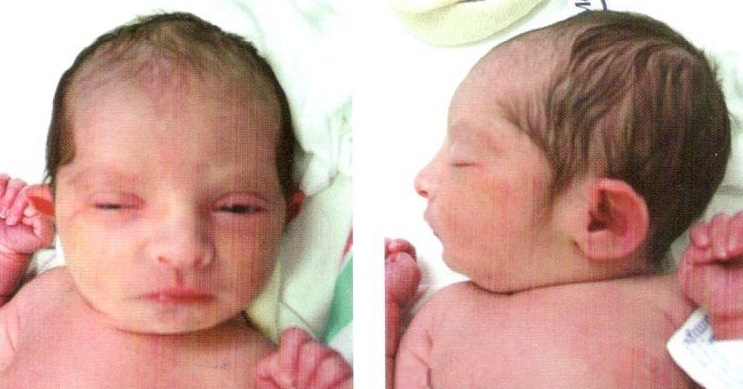
Клиническая картина характеризуется микроцефалией, расщелиной лица, двусторонним расщеплением верхней губы, полным расщеплением неба, маленькими глазными яблоками либо полным их отсутствием, короткой шеей, маленькими деформированными низко расположенными ушами, полидактилией, дистрофическими изменениями ногтей и костного скелета. Отмечаются также пороки развития сердца, желудка, кишечника и других органов.
Синдром трисомии-Х впервые описан в 1959 г. Частота данной патологии составляет среди новорожденных 0,1%, а среди умственно отсталых — 0,6%. Большинство лиц женского пола с трисомией-Х выявляется среди больных психиатрических лечебниц. Клиническая картина характеризуется аномалиями развития скелета, внутренних органов, различными психическими проявлениями и интеллектуальной недостаточностью. Среди полиморфизма признаков трисомии-Х наиболее характерными являются: низкий рост, аномалии ушей, прикуса, высокое стояние твердого неба, короткие пальцы, искривленный мизинец, широкий промежуток между 1 и 2 пальцами на стопах, синдактилия, недоразвитие половых функций.
Умственная отсталость проявляется в виде легкой или умеренной степени. Характерны эмоциональные расстройства (вспыльчивость, агрессивность, неустойчивость настроения и немотивированные поступки). Девочки с синдромом трисомии-Х с трудом, но в большинстве случаев (легкая степень умственной отсталости) обучаются в массовых школах.
К хромосомным синдромам, помимо вышеописанных, относится большая группа так называемых семейных форм умственной отсталости, когда совершенно точно доказано наличие данной патологии у близких родственников.
Синдром Аперта (акроцефалосиндактилия) — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, экзофтальмом, деформацией зубов и синдактилиями. Синдром описан французским педиатром Апертом (Е. Apert) в 1906 г.
Синдром Крузона — наследственное заболевание, характеризующееся умеренной или тяжелой умственной отсталостью, преждевременным срастанием швов черепа, уменьшением мозгового вещества, экзофтальмом, вторичной атрофией зрительных нервов, прямоугольным расположением большого пальца к кисти. Впервые синдром описан французским врачом Крузоном (О. Crouson) в 1912 г.
Синдром Берьесона—Форсмана—Лемана — синдром характеризующийся умственной отсталостью в сочетании с избыточным весом. Впервые описан американскими врачами Берьесоном (М. Berjeson) Форсманом (Н. Foreman) и Леманом (О. Lehman) в 1963 г. Клиническая картина заболевания проявляется выраженным ожирением и прогрессирующей умственной отсталостью. Ожирение носит не равномерный характер. Жир откладывается преимущественно на бедрах, груди и лице, что придает своеобразный вид такому больному (бочкообразная карликовая фигура с заплывшим лицом, большими ушами и узкими разрезами глаз). У больных часто отмечаются эпилептические припадки. Умственная отсталость колеблется от умеренной до тяжелой степени. Данная патология встречается только у лиц мужского пола, но носителями патологического гена являются женщины.
Синдром Прадера—Вилли — наследственное заболевание, характеризующееся глубокой умственной отсталостью, низким ростом, гипогенитализмом, ожирением, резко выраженной мышечной гипотонией.
Синдром Книппеля—Фейля (синдром короткой шеи) — наследственное семейное заболевание, обусловленное врожденными аномалиями развития скелета и внутренних органов в сочетании с тяжелой степенью умственной отсталости. Клиника синдрома подробно описана французскими врачами Клиппелем Фейлем в 1912 г.
Аномалия развития характеризуется следующими проявлениями: короткой шеей как результат количественного уменьшения шейных позвонков, ограничением подвижности головы, расщеплением твердого неба, бочкообразной грудной клеткой, врожденными пороками сердца, добавочными долями или отсутствием отдельных долей легких, синдактилиями (сращение пальцев конечностей), глухотой вследствие заращения наружных слуховых проходов, сужением анального отверстия и многими другими симптомами. Интеллектуальная недостаточность является результатом тяжелой умственной отсталости
Лечение ЗПРР при хромосомных заболеваниях.
Исследования последних десятилетий выявили, что у большинства детей с речевыми и поведенческими проблемами в различной степени нарушены функции мозжечка и базальных ганглиев. Именно функционирование мозжечка определяет успешность ребенка в обучении. С этой целью применяется уникальная разработка Центра авиакосмической медицины — подошвенный имитатор опорной нагрузки «Корвит», применяемый для нейрофизиологической регуляции стато-кинетической функции ЦНС. В основе терапевтического воздействия аппарата «Корвит» лежит процесс активации опорной афферентации, отвечающей за нормализацию процессов возбуждения и торможения в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в головном мозге, способствующих восстановлению координации движений, и, опосредованно, улучшению речи и мышления.
Обязательным звеном в лечебном комплексе у детей с наличием речевых расстройств является занятия с клиническим психологом, а также логопедическая коррекция, которая включает диагностику степени нарушений, ежедневные занятия, направленные на улучшение речевой функции и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
На фоне сочетания проведения биофизической активации со вспомогательными методиками лечения наблюдаются положительные изменения, которые могут быть видны уже через несколько процедур, но максимальный эффект развивается через полтора-три месяца после курса. Как правило, для закрепления полученных результатов и дальнейшего развития двигательных и когнитивных навыков специалистами центра рекомендуется повторный курс лечения через 5-6 месяцев.


